Cystic fibrosis
This article was accepted into the corpus but its outbound wikilinks were never NER-processed — typical at the deepest BFS hop or when the run's entity cap was reached. No expansion funnel to show.
| Cystic fibrosis | |
|---|---|
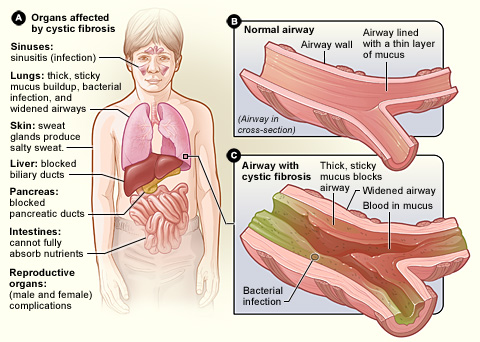 National Heart Lung and Blood Institute, National Institutes of Health (NIH) · Public domain · source | |
| Name | Cystic fibrosis |
| Field | Pulmonology, Gastroenterology, Genetics |
| Symptoms | Chronic cough, recurrent infections, malabsorption, infertility |
| Complications | Bronchiectasis, respiratory failure, diabetes mellitus, liver disease |
| Onset | Childhood |
| Duration | Lifelong |
| Causes | Mutation in a single gene |
| Diagnosis | Sweat test, genetic testing |
| Treatment | Multidisciplinary care, CFTR modulators, airway clearance |
| Frequency | ~1 in 2,500–3,500 live births in populations of European descent |
Cystic fibrosis
Cystic fibrosis is a hereditary multisystem disorder characterized by persistent pulmonary disease, exocrine pancreatic insufficiency, and elevated sweat chloride. It arises from defects in an epithelial ion channel leading to viscous secretions, recurrent infections, and progressive organ damage. Management requires coordinated multidisciplinary care combining pharmacotherapy, physiotherapy, nutrition, and transplantation in advanced disease.
Introduction
Cystic fibrosis was first delineated in the 20th century amid advances in pediatrics and pathology alongside institutions such as Johns Hopkins Hospital and Great Ormond Street Hospital. Early descriptions overlapped with work at Boston Children's Hospital, Massachusetts General Hospital, and laboratories influenced by investigators at Cold Spring Harbor Laboratory and Rockefeller University. Recognition of the genetic basis accelerated with techniques developed at University of Cambridge, Harvard Medical School, and University of California, San Francisco. Major patient organizations including Cystic Fibrosis Foundation and international networks based in London, Toronto, and Sydney have driven research, newborn screening, and advocacy.
Pathophysiology and Genetics
The disorder is caused by mutations in the CFTR gene located on chromosome 7, discovered with methods from Sanger Institute and mapped using linkage strategies employed at Stanford University and MIT. CFTR encodes an ATP-gated chloride channel expressed in epithelia of the airways, pancreas, intestine, and sweat glands; dysregulation parallels findings in ion transport studies from National Institutes of Health and Karolinska Institute. Classifications of CFTR variants (classes I–VI) were refined by research groups at Yale School of Medicine, University of Toronto, and Leiden University Medical Center. The most common pathogenic allele, ΔF508, emerged from population studies performed by teams at Imperial College London and University College London. Cellular consequences—impaired mucociliary clearance, dehydration of airway surface liquid, and altered host defense—reflect mechanistic models developed at Max Planck Society and University of Pennsylvania.
Clinical Manifestations
Pulmonary disease manifests as chronic productive cough, recurrent bacterial infections with pathogens such as Pseudomonas aeruginosa, Staphylococcus aureus, and Burkholderia cepacia complex, and bronchiectasis noted in imaging centers like Mayo Clinic and Cleveland Clinic. Gastrointestinal involvement includes exocrine pancreatic insufficiency, distal intestinal obstruction syndrome, and hepatobiliary disease observed in cohorts studied at King's College Hospital and Great Ormond Street Hospital. Male infertility due to congenital bilateral absence of the vas deferens has been described in literature from University of Oxford and McGill University. Less common presentations include CF-related diabetes, managed using protocols from Joslin Diabetes Center and Karolinska University Hospital.
Diagnosis
Diagnostic algorithms employ newborn screening programs instituted by public health agencies in United States, United Kingdom, Australia, and Canada and utilize immunoreactive trypsinogen assays refined by laboratories at Centers for Disease Control and Prevention and Public Health England. Confirmatory testing includes quantitative pilocarpine iontophoresis sweat testing standardized by clinical laboratories at Mayo Clinic and genetic testing panels developed by companies and centers affiliated with Broad Institute and Genentech. Diagnostic criteria and guidelines have been promulgated by specialist societies including European Respiratory Society, American Thoracic Society, and national CF trusts in Scotland and New Zealand.
Treatment and Management
Therapeutic advances include inhaled mucolytics, airway clearance techniques popularized in physiotherapy programs at Royal Brompton Hospital and SickKids Hospital, and aggressive antimicrobial strategies informed by microbiology labs at Johns Hopkins Hospital and University of Washington. Pancreatic enzyme replacement and nutritional support follow protocols from Children's Hospital of Philadelphia and UCSF Benioff Children's Hospital. CFTR modulators—ivacaftor, lumacaftor, tezacaftor, elexacaftor—were developed through collaborations involving Vertex Pharmaceuticals, academic groups at University of North Carolina, and regulatory evaluations by agencies such as Food and Drug Administration and European Medicines Agency. Advanced lung disease management includes long-term oxygen therapy, noninvasive ventilation, and lung transplantation coordinated by centers like University of Toronto and Cleveland Clinic. Multidisciplinary care models draw on frameworks from National Health Service CF clinics and specialist networks in Ireland.
Prognosis and Epidemiology
Survival and quality of life have improved substantially with newborn screening, antibiotics, nutritional care, and CFTR modulators; longitudinal outcome studies from Cystic Fibrosis Foundation Patient Registry and European Cystic Fibrosis Society document rising median survival comparable to registries maintained at Australian Cystic Fibrosis Data Registry and national databases in Germany. Prevalence varies by ancestry, with highest carrier frequencies reported in populations studied at University of Edinburgh and University of Groningen. Socioeconomic determinants and access to specialty centers such as St. Vincent's Hospital and Royal Children's Hospital influence outcomes; public health initiatives by agencies including World Health Organization and national ministries of health affect newborn screening coverage and transplant availability.
Category:Genetic disorders